FAQ
Sections:
- Successful installations
- 64-bit Linux
- Intel compilers
- Apple Macintosh running Mac OS X
- Optimized BLAS libraries
- Precompiled packages on Ubuntu/Debian
- Compiling LAPACK on RedHat9 exits with an error
- PETSc does not compile on RedHat 6.2 with BOPT=g_c++ or BOPT=O_c++
- DEC/Compaq/hp Alpha machines running OSF/1, Tru64
- magpar crashes with a segmentation violation
- Single precision arithmetics
- Single processor version without MPI
- Compiling magpar in Cygwin for Windows
- Compiling magpar using the MinGW compilers in Cygwin
- Running magpar on Windows
- Graphical User Interface for Windows
- Installing Python
- Additional solvers and libraries for PETSc
- Links to other FAQs, troubleshooting guides
- What is "magpar" worth (at least ;-) ?
- Other micromagnetics software
- Installation of old library versions
- How does boundary matrix size scale with mesh length?
- Interpolating in a tetrahedral mesh with barycentric coordinates
Successful installations
Please send me an email, if you have successfully installed magpar on a system different from those listed below: magpar(at)magpar.net
| CPU | OS | magpar version | comment |
|---|---|---|---|
| various AMD and Intel dual and quad-core processors | CentOS 4.x, 5.x (64 bit) | 0.8-0.9 | using automated installation with Makefile.libs, GCC 4.2.1, and self compiled ATLAS 3.7.36 on 64-bit Linux |
| AMD Opteron Processor 250 (2 x 2.4 GHz) | Red Hat Enterprise Linux 3 (64 bit) | 0.5-0.8 | 64-bit Linux |
| Sun UltraSPARC IV+ | Sun Solaris 10 (64 bit) | 0.8 | InstallationOnSolarisSPARC |
| AMD Athlon64 3200 | Linux (64 bit) | 0.4 | 64-bit Linux |
| Intel Xeon, AMD Athlon MP | RedHat 7.3 | 0.2-0.8 | using RedHat's GNU compilers 2.96, atlas3.6.0_Linux_ATHLON and Intel compilers |
| Pentium M | Debian 4.0 (etch), and Debian testing (lenny/sid) | 0.1-0.9 | using atlas3.6.0_Linux_P4SSE2, GCC 4.1.3, GCC 4.2.1 |
| IBM BlueGene/L | Linux/custom OS | 0.8pre4 | 9-teraflop system; see paper by Biskeborn et al. IEEE Trans. Magn., Vol. 46, No. 3, March 2010, 880 - 885 (Publications) |
| Pentium 4 | RedHat 9 | 0.2-0.9 | using RedHat's RPM packages for LAPACK, libpng, zlib; using MPICH, because mpi++.h is missing in RedHat's LAM/MPI |
| Apple PowerMac G5 | Mac OS X 10.4.2 w/ Xcode 2.1 | 0.5 | powerpc-apple-darwin8-gcc-4.0.0 (GCC) 4.0.0 (Apple Computer, Inc. build 5026) on Apple Macintosh running Mac OS X |
| Apple iBook G4 1.2 GHz | Mac OS X 10.3 | 0.5 | Apple Macintosh running Mac OS X |
| Pentium III | Fedora Core 2 | 0.3-0.5 | LAM/MPI 7.1.1 |
| Alpha EV68 | Tru64 5.1 | 0.1-0.5 | DEC/Compaq/hp Alpha machines running OSF/1, Tru64 : using MPICH, dxml; Compaq AlphaServer SC45 V2.6 consisting of 11 ES45 nodes with 4 processors (1 GHz, 8 MB cache/CPU) each and 16 GB RAM/node, connected with Quadrics Supercomputer World high speed interconnect |
| Alpha EV6 (21264) | Tru64 5.0A | 0.1-0.3 | using MPICH, using dxml |
| Alpha EV56 | RedHat 6.2 | 0.1-0.3 | g++ is broken, compiled PETSc only with BOPT=O (no C++ support, no TAO, no energy minimization), self compiled ATLAS |
| Alpha EV5 (21164) | Tru64 4.0F | 0.1-0.3 | patching of $PETSC_DIR/src/sys/viewer/impls/mathematica necessary: replace snprintf by sprintf (see below: DEC/Compaq/hp Alpha machines running OSF/1, Tru64); using dxml |
| AMD Athlon | RedHat 6.2 | 0.1-0.3 | original RedHat C++ compiler [gcc version egcs-2.91.66 19990314/Linux (egcs-1.1.2 release)] is broken compiled PETSc BOPT=O_c++ with g++ of gcc 3.3.1 (compiled from source) using atlas3.4.1_Linux_ATHLON |
| Intel Xeon 2.4 GHz | Windows XP | 0.5-0.9 | see below: Compiling magpar in Cygwin for Windows, Running magpar on Windows, and Single processor version without MPI |
64-bit Linux
On recent 64-bit Linux distributions, the Installation instructions apply and the installation works out of the box. Still, the following suggestions might be helpful.
Problem
Compilation of magpar and all required libraries on AMD 64-bit processors (Athlon64, Opteron) running 64-bit Linux.
Solution 1
AMD Opteron Processor 250 (2 x 2.4 GHz) running Red Hat Enterprise Linux WS release 3 (Taroon Update 4)
LAPACK: The (incomplete) implementation distributed with ATLAS works: http://prdownloads.sourceforge.net/math-atlas/atlas3.6.0_Linux_HAMMER64SSE2.tar.gz?download
Otherwise use RedHat's LAPACK rpm "lapack-3.0-20" - also available from RPMfind: ftp://rpmfind.net/linux/redhat/enterprise/3/en/os/i386/SRPMS/lapack-3.0-20.src.rpm
The generic LAPACK from netlib does not work!
Everything else works as described in the Installation instructions.
Solution 2
submitted by Richard Boardman (thanks!):
Good news! I've managed to get magpar running on a pure 64-bit Linux system on AMD64. It was a bit of an adventure.
ATLAS: This needed to be built manually, as it looks like they've not used -fPIC (position independent code) everywhere. I needed to get the source and override (despite the warnings) the compiler flags to send -fPIC and -m64 to both the C and the Fortran components (IIRC)
LAPACK: Setting the -fPIC flags and the -m64 thing does the trick, apart from at the link stage. Two files needed to be compiled manually (and then the linking done manually):
g77 -c -m64 -fPIC dlamch.f g77 -c -m64 -fPIC slamch.f
as LAPACK tries to compile these and these alone (weird) without the PIC.
MPI(CH): absolutely must have the -fPIC and the -m64 stuff in there. I think it's pretty much OK to go with:
CFLAGS="-fPIC -m64" FFLAGS="-fPIC -m64" RSHCOMMAND="ssh" \ ./configure && make
though watch out - if PETSc fails, then check carefully where the R_X86_64_32 relocation error occurred and (a) try and make the natural build process cover this, (b) manually build it if this fails. I guess this is true for all of them.
PETSc: The above stages are necessary for PETSc 2.2.(0) - assuming everything above went to plan, then PETSc will build, but make sure the following lines in bmake/linux/variables are set:
C_CC = gcc -fPIC -m64 C_FC = g77 -Wno-globals -fPIC -m64 O_COPTFLAGS = -O -Wall -Wshadow -fomit-frame-pointer -fPIC -m64 O_FOPTFLAGS = -O -fPIC -m64 CXX_CC = g++ -fPIC -m64 CXX_FC = g77 -Wno-globals -fPIC -m64 GCXX_COPTFLAGS = -g -m64 -fPIC OCXX_COPTFLAGS = -O -m64 -fPIC GCOMP_COPTFLAGS = -g -m64 -fPIC OCOMP_COPTFLAGS = -O -m64 -fPIC
Here is a suggestion for (quite pedantic ;-) compile flags for PETSc:
export CFLAGS="-O3 -fPIC -march=k8 -msse2 -m64 -mfpmath=sse -m3dnow -fexpensive-optimizations -fforce-addr -fforce-mem -finline-functions -funroll-loops -Wall -Winline -W -Wwrite-strings -Wno-unused"
zlib, libpng and magpar: These should have -m64 -fPIC in their build process for consistency.
If all this is done, hopefully a nice static pure 64-bit binary should be built, Opteron-friendly :)
Intel compilers
(tested with Intel compiler (icc, ifort) version 9.0)
In order to compile magpar (and all the libraries) with the Intel compilers one can just add the following snippet to the host specific Makefile.in.$HOSTNAME or Makefile.in (update paths to your installation directories!)
CC=/usr/local/bin/icc CPP=/usr/local/bin/icc -E CXX=/usr/local/bin/icpc FC=/usr/local/bin/ifort TIMER=EXT_ETIME # Generate code for Intel Pentium 4 processors and enables new # optimizations in addition to Intel processor-specific optimizations. OPTFLAGS += -O3 -xN # http://icl.cs.utk.edu/lapack-forum/viewtopic.php?t=295 NOOPT = -O3 -fltconsistency PETSC_XTRALIBS=/usr/local/lib/libifcore.a
These settings are required especially for the compilation of LAPACK (set the variables in the make.inc file) as discussed here: http://icl.cs.utk.edu/lapack-forum/viewtopic.php?t=295
It is possible to use the precompiled ATLAS libraries, even though the Intel Math Kernel Library, which implements all BLAS and LAPACK routines, might be a good option, too.
The configuration of PETSc might fail when it tests the linking of Fortran object files with mpicc. To solve this problem it is necessary to add "-lirc" to "MPI_OTHERLIBS" in mpif90.
If you use Makefile.libs to install the libraries, it will use the settings in Makefile.in.$HOSTNAME or Makefile.in.defaults. If you compile by hand, define the variables above on the command line (and export them if necessary).
Apple Macintosh running Mac OS X
Problem
Compilation of magpar and all required libraries on Apple Macintosh running Mac OS X.
Solution 1
submitted by Richard Boardman (thanks!)
tested on Apple iBook G4 1.2 GHz with Mac OS X 10.3
I got everything up and running on Darwin/G4 [OSX to everyone else ;)]; here are a few observations.
1] get Fink* from SourceForge.net: Fink is a Debian-like package manager for Mac OS X, and contains amongst other things ATLAS and LAPACK
2] get ATLAS from Fink
3] get LAPACK from Fink
4] download MPICH and configure as usual I believe LAM/MPI and/or the packages from Fink might work, too.
5] ParMetis-3.1
- one should adjust the include and lib paths in ParMetis' Makefile.in to point to /sw/lib and /sw/include to pick up ATLAS and LAPACK.
- note that on my Panther setup I needed to place /usr/include/malloc in my include path for ParMetis as malloc.h wasn't being picked up by default
- also note that the usual /usr/lib and /usr/include should be left in the Makefile.in
6] SUNDIALS - download and configure as usual
7] PETSc 2.3.0
Copy PETSc-config-magpar.py to darwin-gnu-magpar.py
cp PETSc-config-magpar.py darwin-gnu-magpar.py
update it (Fink installs the libraries, e.g. ATLAS, LAPACK, in /sw) and add the following line to the "configure_options":
'--with-cxx=g++', # mpiCC does not work (some case sensitivity issue)
Then, configure and run as usual.
8] TAO, zlib, and libpng as usual
Solution 2
submitted by Greg Parker (thanks!)
tested on Apple PowerMac G5 2x2.0 GHz with Mac OS X 10.4.1 w/ Xcode 2.1 and
Apple PowerMac G5 2x2.5 GHz with Mac OS X 10.4.1 w/ Xcode 2.0
Turns out things are pretty easy on Tiger. No need for Fink or a Fortran compiler. Apple supplies their own BLAS/LAPACK libraries and headers, so no need for Atlas or Lapack. If they are installed, the configure system of PETSc 2.3.0 finds them by itself.
LAM/MPI: Download lam-7.1.1.dmg.gz from http://www.lam-mpi.org/7.1/download.php and use the installer, which puts it in /usr/local/...
Then type:
cd $PD mkdir mpi cd mpi ln -s /usr/local/bin ln -s /usr/local/include ln -s /usr/local/lib PATH=$PD/mpi/bin:$PATH export PATH lamboot -v
ParMetis 3.1: Edit $MAGPAR_HOME/ParMetis-3.1/Makefile.in (do not modify CC or LD variables as described here: ParMetis )
# INCDIR = -I/usr/local/include -I/usr/include -I/usr/include/malloc # LIBDIR = -L/usr/local/lib -L/usr/lib
and compile as usual.
SUNDIALS 2.1.0: Configure with the following command
./configure --disable-f77 --with-mpi-incdir=$PD/mpi/include --with-mpi-libdir=$PD/mpi/lib
and compile as usual.
PETSc 2.3.0
./config/configure.py --with-fortran=0 --with-mpi-include=$PD/mpi/include \
--with-clanguage=cxx --with-debugging=0 \
--with-mpi-lib=[$PD/mpi/lib/libmpi.a,$PD/mpi/lib/liblammpi++.a,$PD/mpi/lib/liblam.a] \
--with-cxx=mpic++ -CXXOPTFLAGS="-O3 -Wno-long-double" --with-mpirun=mpirun
PETSC_ARCH=<arch> # where <arch> is whatever config said, e.g. darwin8.2.0
export PETSC_ARCH
PETSC_DIR=$PWD
export PETSC_DIR
make all
make test # be sure to have X11 running first, if it fails, may have to type 'lamboot -v' again
libpng-1.2.5
cp scripts/makefile.macosx ./makefile make # you will get errors compiling pngtest, but you can ignore them
Compiling magpar itself
In Makefile.in change the PETSc architecture to whatever PETSc's config said above
PETSC_ARCH = <arch> CFLAGS += -O2 -faltivec
Optimized BLAS libraries
It is highly recommended to use any machine specific (vendor provided and highly optimized) libraries. On most high performance machines there are optimized BLAS and LAPACK libraries available, e.g.:
In addition, the following implementations are available:
- ATLAS: Automatically Tuned Linear Algebra Software - recommended
- Generic BLAS and LAPACK implementations from netlib - portable but slow
- BLAS by Kazushige Goto: Visiting Scientist, FLAME project, UT-Austin - untested
- Intel Math Kernel Library: Optimized library for Intel Intel processors - untested
- AMD Math Core Library: Optimized library for AMD processors - untested
Precompiled packages on Ubuntu/Debian
Using Debian-based distributions (including Ubuntu) one can save a lot of time by using some of the pre-built packages.
The following instructions pertain specifically to Debian Lenny and were used successfully in June 2008 and saved in Makefile.in.host_debian, which can serve as a template for your Makefile.in.$HOSTNAME.
PETSc and Sundials still have to be compiled from source, since the Debian packages were built for uniprocessor systems only and do not work with MPICH. TAO also must be built from source. For this purpose one can still use the Automated Installation for the individual libraries like this
cd $MAGPAR_HOME/src make -f Makefile.libs sundials petsc tao
or install them using the Manual Installation method.
Thanks to Daniel Lenski for his installation report.
Compiling LAPACK on RedHat9 exits with an error
Problem
The GNU Fortran compiler shipping with RedHat 9 (GNU Fortran (GCC 3.2.2 20030222 (Red Hat Linux 3.2.2-5))) has problems with complex variables.
... SEP: Testing Symmetric Eigenvalue Problem routines ./xeigtstc < sep.in > csep.out 2>&1 make: *** [csep.out] Error 139
At this stage, the LAPACK library has been compiled successfully, but the complex test programs fail.
Solution
Since magpar does not use any complex variables, one can safely ignore this error message and continue with the installation.
PETSc does not compile on RedHat 6.2 with BOPT=g_c++ or BOPT=O_c++
Problem
The GNU C++ compiler shipping with RedHat 6.2 (gcc version egcs-2.91.66 19990314/Linux (egcs-1.1.2 release)) is broken and fails with an internal error:
libfast in: /home/scholz/work/magpar/libs/petsc-2.1.6/src/vec/esi eindexspace.c: In method `::esi::petsc::::esi::petsc::IndexSpace<int>:: IndexSpace(class ::esi::::esi::IndexSpace<int> &)': eindexspace.c:21: Internal compiler error. ...
Solution
GCC 2.95.3 (shipping with SuSE 8.0), GCC 2.95.4 (shipping with Debian 3.0 woody) and GCC 3.2.2 have proved to work. Get some binary package for your platform or compile GCC from source.
You can get the latest GCC release from here:
http://gcc.gnu.org/
http://gcc.gnu.org/install/binaries.html
http://gcc.gnu.org/mirrors.html
DEC/Compaq/hp Alpha machines running OSF/1, Tru64
Problems linking various libraries with "ar"
Problem
The following error occured on a Compaq machine running Tru64 UNIX V5.1A (Rev. 1885):
/: write failed, file system is full ar: error writing archive member contents: [...] *** Exit 1 Stop.
after compiling the source files of MPICH (and ParMetis-3.1, too) when "make" tried to create the libraries using "ar".
Solution
Set the environment variable "TMPDIR" to a directory/partition with sufficent free space, e.g. your home directory:
TMPDIR=$HOME export TMPDIR
and recompile.
Problems with snprintf
Tru64 version 4.x does not provide an "snprintf" function. Thus, the compilation of PETSc fails in $PETSC_DIR/src/sys/src/viewer/impls/mathematica/. A simple replacement of snprintf by sprintf and omitting the size argument solves the problem.
in $PETSC_DIR/src/sys/src/viewer/impls/mathematica/mathematica.c
replace:
snprintf(linkname, 255, "%6d", ports[rank]);
with:
sprintf(linkname, "%6d", ports[rank]);
and all other occurences of snprintf in this file accordingly.
Compiling ParMetis
If you are using the ELAN libraries (for Quadrics high speed interconnect) instead of MPI(CH) over Ethernet:
# modify $MAGPAR_HOME/src/Makefile.in to use # the normal C compiler instead of mpicc CC = cc XTRALIBS = -lmpi -lelan LD = cc # test example prun -p scp1 -n 4 -s -o out -e err ./ptest rotor.graph
Otherwise use a "normal" MPI library (MPICH, LAM/MPI) and compile as usual.
Compiling SUNDIALS version 2.1
If you are using the ELAN libraries (for Quadrics high speed interconnect) instead of MPI(CH) over Ethernet:
# use the normal C and Fortran compilers instead of mpicc and mpif77 ./configure --with-mpicc=cc --with-mpif77=f77
Compiling SUNDIALS version 1.0
cd $PD/sundials
# replace compiler "gcc" by "cc"
# remove compiler option "-ffloat-store"
files=`find . -name "*"`
for i in `grep -l "CFLAGS =" $files`; do
sed "s/gcc/cc/g" $i | sed "s/-Wall -ffloat-store//g" > $i.tmp
mv $i.tmp $i
done
# optional: compile with optimizations
# (no improvement/speedup found)
for i in `grep -r -l "CFLAGS =" $files`; do
sed "s/CFLAGS = /CFLAGS = -O3 /g" $i > $i.tmp
mv $i.tmp $i
done
unset files
Compiling PETSc 2.3.0
Install Python (if necessary) as described in the Installing Python.
Modify $MAGPAR_HOME/src/PETSc-config-magpar.py:
... # use Compaq Extended Math Library (CPML/CXML) '--with-blas-lib=libcxml', # use ELAN libraries (if you have Quadrics high speed interconnect) '--with-mpi-lib=libmpi.a,libelan.a', ...
Otherwise use a "normal" MPI library (MPICH, LAM/MPI) and compile as usual.
Compiling PETSc 2.2.1 and earlier
...
PETSC_DIR=$PD/petsc-2.2.1
export PETSC_DIR
# choose suitable predefined configuration (directory name) for your platform
PETSC_ARCH=alpha
export PETSC_ARCH
cd ..
ln -s predefined/$PETSC_ARCH
cd $PETSC_ARCH
cp packages packages.bak
# edit the file "packages":
# Location of BLAS and LAPACK
# BLASLAPACK_LIB = -ldxml
# use ELAN libraries (if you have Quadrics high speed interconnect)
# MPI_LIB = -L${MPI_HOME}/lib -lmpi -lelan
# otherwise use MPICH (as usual)
...
cp petscconf.h petscconf.h.bak
# edit petscconf.h if you want to have static binaries (recommended):
# replace "#define PETSC_USE_DYNAMIC_LIBRARIES 1"
# by "#undef PETSC_USE_DYNAMIC_LIBRARIES"
# add "#define PETSC_HAVE_NETDB_H"
# (if you really have a "netdb.h" somewhere in /usr/include)
#
...
Compiling libpng
... cp scripts/makefile.dec Makefile # edit the Makefile if necessary: # update the paths to zlib: # ZLIBLIB=../zlib-1.1.4 -lz -lm # ZLIBINC=../zlib-1.1.4 make make test
Compiling magpar
Update Makefile.in if you are using PETSc 2.2.1 or earlier:
... # set the proper PETSC_ARCH (which was used for compiling PETSc): # PETSC_ARCH = alpha # link statically: # CLINKER_STATIC = -non_shared # optional: really tough checking with Tru64's cc: # CFLAGS += -portable -check -verbose ...
magpar crashes with a segmentation violation
Problem
This segmentation fault might happen during mesh refinement, mesh partitioning (or really just the first call to a METIS function). In parteleser.c, for example, METIS_MeshToDual is called to convert the mesh into its dual graph and obtain the adjacency structure of the mesh. If you happen to have a very weird mesh in which many elements share a single node, some static arrays in Metis are too small.
Solution
You can either generate a better mesh (if this problem occurs many elements are terribly degenerate with high aspect ratio and low quality factor anyway) or patch METIS:
Edit $PD/ParMetis-3.1.1/METISLib/mesh.c:79 and increase the array size:
idxtype *mark, ind[500], wgt[500];
Then you have to recompile the ParMetis package and recompile magpar:
cd $PD/ParMetis-3.1.1/ make cd $MAGPAR_HOME/src/ # remove the magpar binary, so a new one is linked rm magpar.exe make
Single precision arithmetics
NB: magpar does not appear to work in single precision mode at all right now! There seem to be big problems with the KSP and TAO solvers (convergence issues!?).
To compile magpar with single precision floating point arithmetics:
set floating point precision in Makefile.in.$HOSTNAME
PRECISION=single
patch TAO to make it compile with PETSc in single precision (patch not available yet)
recompile PETSc, Sundials, TAO
make -f Makefile.libs petsc tao sundials
recompile magpar
make
Single processor version without MPI
Compile magpar according to the Installation instructions with the following modifications:
ParMetis
In $PD/ParMetis-3.1.1/METISLib/metis.h remove line 25:
#include "../parmetis.h" /* Get the idxtype definition */
Compile serial version only:
cd METISLib make CC=gcc LD=gcc
Do not use Metis-4.0.1 (November 1998), because it contains a couple of bugs, which have been ironed out in the Metis version included in ParMetis!
SUNDIALS
./configure --prefix=$PWD --disable-mpi make make -i install
PETSc
modify $MAGPAR_HOME/src/PETSc-config-magpar.py:
comment out all MPI related lines except for:
'--with-mpi=0',
Then configure and compile as usual.
Compiling magpar in Cygwin for Windows
Here is a recipe for compiling magpar in the Cygwin environment on a Windows machine:
- get Cygwin: http://cygwin.com/
- download the setup program http://cygwin.com/setup.exe and run it
- not all packages are required - here is an overview of recommended packages:
Category Package ------------------------------------------------ Admin none Archive none Audio none Base all Database none Devel binutils, gcc, gcc-core, gcc-g++, gcc-g77, gcc-mingw-*, make Doc none (recommended: cygwin-doc, man) Editors none Games none Gnome none Graphics libpng12* Interpreters gawk, python, perl Libs zlib Mail none Math lapack Mingw none Net none Publishing none Shells bash, (recommended: mc) Text less Utils bzip2, cygutils, diffutils (required by PETSc) Web wget X11 none
In Makefile.in.defaults or your own Makefile.in.$HOSTNAME set
ATLAS_DIR=/usr/lib
to use Cygwin's LAPACK.
Follow the general Installation instructions and those for Single processor version without MPI. If magpar is compiled in the Cygwin environment, magpar will generate complete inp files (cf. project.INP.inp).
In addition, users have reported successful parallel magpar installations/runs with MPICH in the Cygwin environment:
- Download and install the Microsoft Visual C++ 2005 SP1 Redistributable Package (required by MPICH Windows binary packages).
- Download and install a suitable MPICH Windows binary package (*.msi) from the MPICH2 Downloads page.
- Update the MPI_DIR variable in your magpar Makefile.in.$HOST or Makefile.in.defaults to the MPICH2 installation directory, e.g. "C:\Program Files\MPICH2\".
- Follow the general instructions for compilation in Cygwin above.
- Run a magpar example with 2 magpar processes on the local machine (e.g. a dual core processor) with a command like this:
mpiexec.exe -n 2 -localonly ./magpar.exe
- Running parallel magpar processes on remote machines should only be a matter of a proper MPICH2 network installation. This requires the installation of MPDs (MPICH daemons) as a service on the remote machines.
magpar executables for Windows and the source code of all required libraries are available on the magpar homepage.
Compiling magpar using the MinGW compilers in Cygwin
This section describes the procedure for compiling magpar using the MinGW compilers in the Cygwin environment on a Windows machine. The advantage over compilation with the native Cygwin compilers is, that the MinGW compiled executables do not require the cygwin.dll any more and the other libraries (see Running magpar on Windows) are linked statically into the executable. Compilation in the native MinGW/MSYS environment does not work (easily), because of PETSc using Python for its configuration.
Currently, the Windows version of magpar has only been compiled and tested in serial model (without MPI support). Thus, the installation procedure described in Single processor version without MPI should be used with the following modifications:
Install Cygwin as described in Compiling magpar in Cygwin for Windows with the addition of packages of the MinGW compilers (included in category "Devel"). The package names are gcc-mingw, gcc-mingw-core, gcc-mingw-g++, gcc-mingw, gcc-mingw-g77.
Compile the required libraries with the following changes (the most important being the addition of the "-mno-cygwin" option for the compilers):
The following steps are automated in the following targets in Makefile.libs :
- lapack_mingw
- parmetis_mingw
- sundials_mingw
- petsc_mingw
- zlib_mingw
- libpng_mingw
BLAS/LAPACK:
cp make.inc.example make.inc # generate BLAS and LAPACK libraries (no optimized BLAS for now) make FORTRAN=g77 LOADER=g77 TIMER=EXT_ETIME OPTS="-funroll-all-loops -O3 -mno-cygwin" blaslib lapacklib cp blas_LINUX.a libblas.a cp lapack_LINUX.a liblapack.a
ParMetis:
make CC=gcc LD=gcc CFLAGS="-O3 -I. -mno-cygwin"
Sundials:
./configure --prefix=$PWD --disable-mpi --with-cflags=-mno-cygwin --with-ldflags=-mno-cygwin
PETSc:
cd $PETSC_DIR export PRECISION=double export OPTFLAGS="-mno-cygwin" export ATLAS_DIR=/usr/lib export PETSC_XTRALIBS="" ./config/PETSc-config-magpar.py
After running ./config/PETSc-config-magpar.py remove the following flags from $PETSC_DIR/PETSc-config-magpar/include/petscconf.h (or $PETSC_DIR/bmake/PETSc-config-magpar/petscconf.h for PETSc 2.3.x):
PETSC_HAVE_GETPAGESIZE PETSC_HAVE_IEEEFP_H PETSC_HAVE_NETDB_H PETSC_HAVE_PWD_H PETSC_HAVE_SYS_PROCFS_H PETSC_HAVE_SYS_RESOURCE_H PETSC_HAVE_SYS_TIMES_H PETSC_HAVE_SYS_UTSNAME_H
simple sed script:
hfile=bmake/PETSc-config-magpar/petscconf.h; \ cp $hfile $hfile.bak; \ cat $hfile.bak | \ sed "/PETSC_HAVE_GETPAGESIZE/,+2 d" | \ sed "/PETSC_HAVE_NETDB_H/,+2 d" | \ sed "/PETSC_HAVE_PWD_H/,+2 d" | \ sed "/PETSC_HAVE_SYS_PROCFS_H/,+2 d" | \ sed "/PETSC_HAVE_SYS_RESOURCE_H/,+2 d" | \ sed "/PETSC_HAVE_SYS_TIMES_H/,+2 d" | \ sed "/PETSC_HAVE_SYS_UTSNAME_H/,+2 d" \ > $hfile
zlib:
make CFLAGS="-O -mno-cygwin"
libpng:
# make sure $PD is set correctly lib=libpng-1.2.33 # adjust to your version of libpng zlib=zlib-1.2.3 # adjust to your version of zlib ./configure --prefix=$PD/$lib --enable-shared=no CFLAGS="-I$PD/$zlib -mno-cygwin" LDFLAGS="-L$PD/$zlib -mno-cygwin" 2>&1 | tee configure.log make && make install && make check
magpar:
# a simple "make" will compile and link the magpar executable make
After compiling magpar (or really any program) one can check which DLLs the program depends on using
objdump -p magpar.exe
see also: http://www.delorie.com/howto/cygwin/mno-cygwin-howto.html
Running magpar on Windows
To run magpar on Windows either check Compiling magpar in Cygwin for Windows or download the archives containing the precompiled binaries from the magpar download page.
One zip archive contains the (executable) program itself and the required libraries:
- magpar.exe: magpar executable
- cygwin1.dll: Cygwin library
- cygblas.dll: BLAS library
- cyglapack.dll: LAPACK library
- cygpng12.dll: PNG library
- cygz.dll: zlib library
To run a magpar simulation
- extract the files from the archive
- copy the magpar executable into a simulation directory (e.g. one of the examples provided separately)
- copy the DLLs to C:\Windows\ (once) or also into each simulation directory
- double click on magpar.exe in the simulation directory
- or run the executable in a "Command Prompt" window (select "Start/Programs/Accessories/Command Prompt" or do "Run.../cmd").
- ideally run the executable in a "Command Prompt" like this "./magpar.exe > stdout.txt" so the informational output of magpar is saved in the file stdout.txt. Then inspect the file stdout.txt as the simulation runs (e.g. with WordPad - not Notepad due to Unix newlines)
The precompiled magpar executable provided
- is only a serial version (it does not support parallelization on SMP machines or clusters)
- generates complete project.INP.inp files
- calculates exchange field and energy separately from anisotropy
- includes K_2 in the uniaxial and cubic anisotropy field and energy
Graphical User Interface for Windows
The Windows executables of magpar are simple programs which just run in a terminal window without any nice user interface. However, thanks to the work of Tomasz Blachowicz and Bartlomiej Baron there is now also a nice graphical user interface for magpar available (see Tomasz's webpage).
Here is a quick introduction how to use it:
- Download MagParExt from its homepage and install it.
- Copy magpar.exe and the DLLs into a project (e.g. example) directory
- Launch MagParExt
- assign a new name to your project (independent of the magpar project/simName)
- Point MagParExt to the project directory using the "Project files/Browse..." button
- Check/modify the simulation parameters using the "Simulation/Configure..." button
- Check/modify the material parameters using the "Materials/Configure..." button
- Configure the application path to magpar.exe using "Options/General"
- If a copy of the Cygwin DLLs is installed in "C:\Windows" there is no need to copy them into each project diretory.
- Run the simulation using the "Simulation/Run" button
- When the simulation finishes import the output data using the "Output data/Import from file..." button (make sure you open the *.log file in the correct directory)
- Click on "Output data/Preview" to have a look at the log file
- Visualize the results using "Graphs"
- Add "Add new graph..." and assign a name to the graph
- Set up "Data Series" by selecting data for x- and y-axis and click "Add"
- Click "Ok" to view the plot
- Go to "Animations" and press play to view them.
References:
See the paper by Tomasz Blachowicz and Bartlomiej Baron in the list of Publications and online at [ arXiv ].
Installing Python
If you do not have Python installed on your system or your Python version is older than 2.2. then download the latest source package of Python, configure, compile, and install it with
./configure --prefix=$PD/python make make install
and update $MAGPAR_HOME/src/PETSc-config-magpar.py with the full path to the python binary, e.g.
#!/home/scholz/work/magpar/libs/python/bin/python
(the environment variable "$MAGPAR_HOME" does not work here!)
Additional solvers and libraries for PETSc
These libraries are optional and they make additional preconditioners/linear solvers available to PETSc. They are not required for magpar.
Please consult the PETSc manual on how to make use of these additional solvers.
hypre
This library is optional and makes additional preconditioners/linear solvers available to PETSc. It is not required for magpar.
Please consult the PETSc manual on how to make use of these additional solvers.
cd $PD
wget http://www.llnl.gov/CASC/hypre/download/hypre-1.8.2b.tar.gz
tar xzvf hypre-1.8.2b.tar.gz
cd hypre-1.8.2b/src
# for MPICH
./configure --with-mpi-include=${MAGPAR_HOME}/libs/mpich/include \
--with-mpi-libs="mpich pmpich fmpich" \
--with-mpi-lib-dirs=${MAGPAR_HOME}/libs/mpich/lib \
--with-blas="-I${MAGPAR_HOME}/libs/atlas/include \
-L${MAGPAR_HOME}/libs/atlas/lib -llapack -lf77blas -latlas -lg2c"
# for LAM/MPI
./configure --with-mpi-include=${MAGPAR_HOME}/libs/lam/include \
--with-mpi-libs="mpi lamf77mpi lam" \
--with-mpi-lib-dirs=${MAGPAR_HOME}/libs/lam/lib \
--with-blas="-I${MAGPAR_HOME}/libs/atlas/include \
-L${MAGPAR_HOME}/libs/atlas/lib -llapack -lf77blas -latlas -lg2c"
# fix utilities/fortran.h (remove one underscore at end of line)
# line 31: define hypre_NAME_C_CALLING_FORT(name,NAME) name##_
# line 32: define hypre_NAME_FORT_CALLING_C(name,NAME) name##_
make
SuperLU
This library is optional and makes additional preconditioners/linear solvers available to PETSc. It is not required for magpar.
Please consult the PETSc manual on how to make use of these additional solvers.
cd $PD
wget http://crd.lbl.gov/~xiaoye/SuperLU/superlu_dist_2.0.tar.gz
tar xzvf superlu_dist_2.0.tar.gz
cd SuperLU_DIST_2.0
# edit make.inc:
# PLAT = _linux
# DSuperLUroot = ${MAGPAR_HOME}/libs/SuperLU_DIST_2.0
# BLASLIB = -L${MAGPAR_HOME}/libs/atlas/lib -llapack -lf77blas -latlas -lg2c
# for MPICH
# MPILIB = -L${MAGPAR_HOME}/libs/mpi/lib -lmpich -lpmpich
# for LAM/MPI
# MPILIB = -L${MAGPAR_HOME}/libs/mpi/lib -lmpi -llamf77mpi -llam
# CC = ${MAGPAR_HOME}/libs/mpi/bin/mpicc
# CFLAGS = -O3
# FORTRAN = ${MAGPAR_HOME}/libs/mpi/bin/mpif77
# FFLAGS = -O3
# LOADER = ${MAGPAR_HOME}/libs/mpi/bin/mpicc
# LOADOPTS = #
# CDEFS = -DAdd_
Links to other FAQs, troubleshooting guides
An extensive list of known problems and difficulties is listed on the PETSc website:
http://www.mcs.anl.gov/petsc/petsc-as/documentation/troubleshooting.html
http://www.mcs.anl.gov/petsc/petsc-as/documentation/faq.html
What is "magpar" worth (at least ;-) ?
Total Physical Source Lines of Code (SLOC) = 11,632 Development Effort Estimate, Person-Years (Person-Months) = 2.63 (31.56) (Basic COCOMO model, Person-Months = 2.4 * (KSLOC**1.05)) Schedule Estimate, Years (Months) = 0.77 (9.28) (Basic COCOMO model, Months = 2.5 * (person-months**0.38)) Estimated Average Number of Developers (Effort/Schedule) = 3.40 Total Estimated Cost to Develop = $ 355,287 (average salary = $56,286/year, overhead = 2.40).
generated using David A. Wheeler's SLOCCount version 2.26
Other micromagnetics software
Here are a few links to other free and commercial micromagnetics packages:
- OOMMF (Object Oriented MicroMagnetic Framework)
- nmag - a flexbile micromagnetic simulation package
- JaMM - Java MicroMagnetics
- RKMAG
- LLG Micromagnetics Simulator
- MicroMagus
- MagOasis
- FEMME - multiscale finite element micromagnetic package
- PC Micromagnetics Simulator
- AlaMag
Installation of old library versions
MPICH1
cd $PD wget ftp://ftp.mcs.anl.gov/pub/mpi/mpich.tar.gz tar xzvf mpich.tar.gz cd mpich-1.2.6 # patch -p0 < ../patchfile # if you experience problems applying the patch # go back to the original mpich.tar.gz ./configure make make testing # Problems with RedHat 6.2 bash: testing fails # solution: replace bash version 1 with version 2: # cd /bin; mv bash bash.bak; ln -s bash2 bash # # install the binaries in $PD/mpich ./bin/mpiinstall -prefix=$PD/mpich # set symbolic link to MPICH installation directory cd $PD ln -s mpich mpi
LAM/MPI
cd $PD wget http://www.lam-mpi.org/download/files/lam-7.1.2.tar.gz tar xzvf lam-7.1.2.tar.gz cd lam-7.1.2 ./configure --prefix=$PD/lam make make install # set symbolic link to LAM installation directory cd $PD ln -s lam mpi # start LAM universe on local machine: lamboot -v
How does boundary matrix size scale with mesh length?
Since the boundary matrix contains one row and one column for every surface node, it is clear that 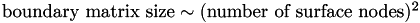 .
.
The surface of the mesh consists of triangles, edges, and points:
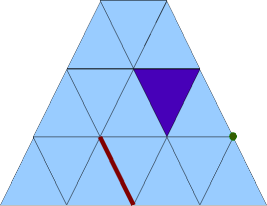
2D surface of a 3D tetrahedral mesh
Their numbers are:
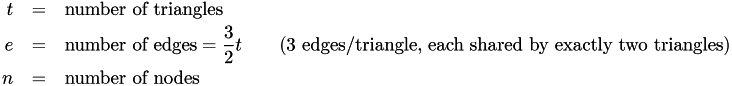
If the mesh is a continuous, perfect simple hexagonal mesh, then each node is shared by six triangles. There are three nodes per triangle and thus:

Consider the process of subdividing every triangle into four smaller triangles. This is what Magpar does when the -refine 1 option is given, and is effectively the same as reducing the mesh length by a factor of 2:
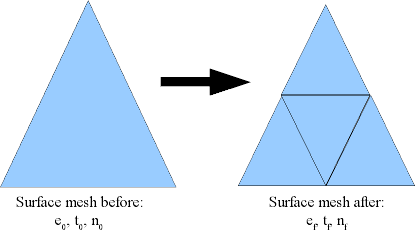
The effect of -refine 1 on a surface mesh
How are 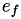 ,
, 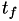 ,
, 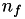 related to
related to 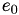 ,
, 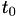 ,
, 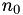 ?
?
-
every triangle becomes four new triangles, thus
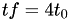
-
a new node is placed along every edge, thus
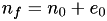
Since 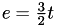 , we get
, we get 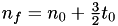 .
.
In the case of a simple hexagonal mesh: 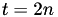 , thus
, thus 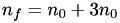 , or
, or 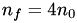 . In this case,
. In this case, 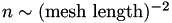 .
.
This is neither an upper bound nor a lower bound, since it is possible for there to be more than 6 triangles sharing each surface on average, or less than 6 on average. But most of the non-hexagonal cases I can think of are pretty pathological, and a quick glance at a mesh produced by Gmsh shows that surface nodes overwhelmingly are shared by 6 triangles.
From this we conclude that to an excellent approximation. Since 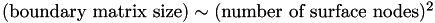 , we conclude that:
, we conclude that: 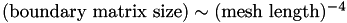
Interpolating in a tetrahedral mesh with barycentric coordinates
Barycentric coordinates are ideal for interpolation within a tetrahedral finite-element mesh (such as used by Magpar). Barycentric interpolation expresses the interpolated function as a weighted sum of the function's values at the four vertices of the tetrahedral cell in which the interpolation point lies. Basically, it assumes linearity of the function within the cell:
![\[ f(\vec{x}) = f \left( \sum_{k=1}^4 \lambda_k \vec{v}_k \right) = \sum_{k=1}^4 \lambda_k f(\vec{v}_k) \quad\forall\; \vec{x} \in \textrm{ the tetrahedral volume with vertices } \vec{v}_1 ... \vec{v}_4 \]](form_19.png)
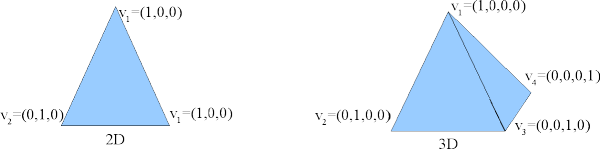
Barycentric coordinates on 2D and 3D simplices (triangles and tetrahedra)
From this basic description, we infer:
-
The sum of the
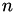 barycentric coordinates, denoted
barycentric coordinates, denoted 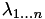 , is exactly 1.
, is exactly 1. -
For any point on the surface/perimeter,
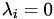 for at least one
for at least one 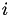 .
. -
For any point within the volume/area,
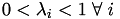 .
.
Given the linearity of this coordinate system, there must be a matrix 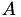 that will transform from barycentric coordinates to Cartesian coordinates, that is
that will transform from barycentric coordinates to Cartesian coordinates, that is 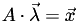 . For the 3D case,
. For the 3D case, 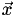 is 3D and
is 3D and 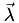 is 4D, thus is 3×4.
is 4D, thus is 3×4.
We know how A maps the tetrahedron's vertices to barycentric coordinates:
![\[ A \left( \begin{array}{c}1\\0\\0\\0\end{array} \right) = \vec{v}_1, \quad A \left( \begin{array}{c}0\\1\\0\\0\end{array} \right) = \vec{v}_2, \quad A \left( \begin{array}{c}0\\0\\1\\0\end{array} \right) = \vec{v}_3, \quad A \left( \begin{array}{c}0\\0\\0\\1\end{array} \right) = \vec{v}_4, \quad \]](form_29.png)
![\[ \textrm{thus} \quad AI = A = \left(\begin{array}{cccc} v_{1x} & v_{2x} & v_{3x} & v_{4x} \\ v_{1y} & v_{2y} & v_{3y} & v_{4y} \\ v_{1z} & v_{2z} & v_{3z} & v_{4z} \end{array}\right) \]](form_30.png)
So far so good, we can map barycentric coordinates to tetrahedral vertices. However, the reverse isn't there yet. We need to recall the additional constraint on the sum of the barycentric coordinates:
![\[\sum_{k=1}^4 \lambda_k = 1\]](form_31.png)
We can combine this with the matrix to get:
![\[ T\vec\lambda = \left(\begin{array}{c}x\\y\\z\\1\end{array}\right) \quad\textrm{where}\quad T=\left(\begin{array}{cccc} v_{1x} & v_{2x} & v_{3x} & v_{4x} \\ v_{1y} & v_{2y} & v_{3y} & v_{4y} \\ v_{1z} & v_{2z} & v_{3z} & v_{4z} \\ 1 & 1 & 1 & 1 \end{array}\right), \quad\textrm{and thus}\quad \vec\lambda=T^{-1}\left(\begin{array}{c}x\\y\\z\\1\end{array}\right) \]](form_32.png)
Now the inverse matrix, 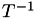 , maps from Cartesian coordinates to barycentric coordinates. Here's the complete algorithm to do barycentric interpolation of a point
, maps from Cartesian coordinates to barycentric coordinates. Here's the complete algorithm to do barycentric interpolation of a point 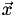 within a tetrahedral mesh:
within a tetrahedral mesh:
-
Compute the barycentric coordinate transformation matrices
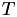 for all the (nearby) cells in the mesh, and invert them to get .
for all the (nearby) cells in the mesh, and invert them to get . -
For each cell and interpolation point, compute
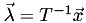 .
. -
For each
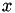 , there will only be at most one such that
, there will only be at most one such that 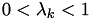 for
for 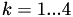 . If point lies on the surface of a tetrahedron, there may be several choices with
. If point lies on the surface of a tetrahedron, there may be several choices with 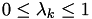 . Pick any one.
. Pick any one. -
Now we know (a) which cell
is in, (b) its barycentric coordinates relative to that cell, and (c) the vertices 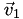 ,
, 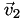 ,
, 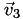 , and
, and 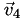 of that cell. Now it's easy: the interpolated function at point is simply the -weighted average of the function's value at the vertices,
of that cell. Now it's easy: the interpolated function at point is simply the -weighted average of the function's value at the vertices,![\[ f_\textrm{interp}(\vec x)= \sum_{k=1}^{4} \lambda_k f(\vec v_k) \]](form_45.png)